
BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains
2020-09-11 | 药学院英文网
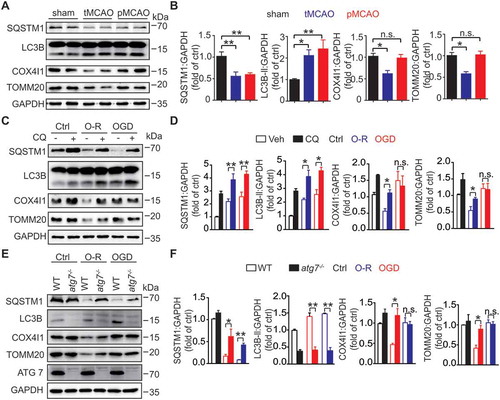
Mitophagy, the elimination of damaged mitochondria through autophagy, promotes neuronal survival in cerebral ischemia. Previous studies found deficient mitophagy in ischemic neurons, but the mechanisms are still largely unknown. We determined that BNIP3L/NIX, a mitophagy receptor, was degraded by proteasomes, which led to mitophagy deficiency in both ischemic neurons and brains. BNIP3L exists as a monomer and homodimer in mammalian cells, but the effects of homodimer and monomer on mitophagy are unclear. Site-specific mutations in the transmembrane domain of BNIP3L (S195A and G203A) only formed the BNIP3L monomer and failed to induce mitophagy. Moreover, overexpression of wild-type BNIP3L, in contrast to the monomeric BNIP3L, rescued the mitophagy deficiency and protected against cerebral ischemic injury. The macroautophagy/autophagy inhibitor 3-MA and the proteasome inhibitor MG132 were used in cerebral ischemic brains to identify how BNIP3L was reduced. We found that MG132 blocked the loss of BNIP3L and subsequently promoted mitophagy in ischemic brains. In addition, the dimeric form of BNIP3L was more prone to be degraded than its monomeric form. Carfilzomib, a drug for multiple myeloma therapy that inhibits proteasomes, reversed the BNIP3L degradation and restored mitophagy in ischemic brains. This treatment protected against either acute or chronic ischemic brain injury. Remarkably, these effects of carfilzomib were abolished in bnip3l -/- mice. Taken together, the present study linked BNIP3L degradation by proteasomes with mitophagy deficiency in cerebral ischemia. We propose carfilzomib as a novel therapy to rescue ischemic brain injury by preventing BNIP3L degradation.

NEWS
-
16
2026.05
-
20
2026.04
-
30
2026.03
-
10
2025.12
-
08
2026.03
-
27
2025.11